Input data and parameters
QualiMap command line
| qualimap bamqc -bam ../mtuberculosis2/2-alignment/pathogen/bam/mycobacteriumTuberculosis/SRR25792495_hisat2.bam -nw 400 -hm 3 |
Alignment
| Command line: | "/home/dguevara/pipeline/src/../tools/hisat2-2.1.0/hisat2-align-s --wrapper basic-0 -x ../mtuberculosis2/2-alignment/pathogen/indices/hisat2/mycobacteriumTuberculosis -S ../mtuberculosis2/2-alignment/pathogen/sam/mycobacteriumTuberculosis/SRR25792495_hisat2.sam -p 16 -1 ../mtuberculosis2/2-alignment/host/fastq/SRR25792495_1.fastq -2 ../mtuberculosis2/2-alignment/host/fastq/SRR25792495_2.fastq" |
| Draw chromosome limits: | no |
| Analyze overlapping paired-end reads: | no |
| Program: | hisat2 (2.1.0) |
| Analysis date: | Thu Feb 01 04:30:22 CST 2024 |
| Size of a homopolymer: | 3 |
| Skip duplicate alignments: | no |
| Number of windows: | 400 |
| BAM file: | ../mtuberculosis2/2-alignment/pathogen/bam/mycobacteriumTuberculosis/SRR25792495_hisat2.bam |
Summary
Globals
| Reference size | 4,411,532 |
| Number of reads | 34,732,954 |
| Mapped reads | 29,829,027 / 85.88% |
| Unmapped reads | 4,903,927 / 14.12% |
| Mapped paired reads | 29,829,027 / 85.88% |
| Mapped reads, first in pair | 14,639,335 / 42.15% |
| Mapped reads, second in pair | 15,189,692 / 43.73% |
| Mapped reads, both in pair | 29,467,908 / 84.84% |
| Mapped reads, singletons | 361,119 / 1.04% |
| Read min/max/mean length | 100 / 100 / 100 |
| Duplicated reads (estimated) | 29,638,321 / 85.33% |
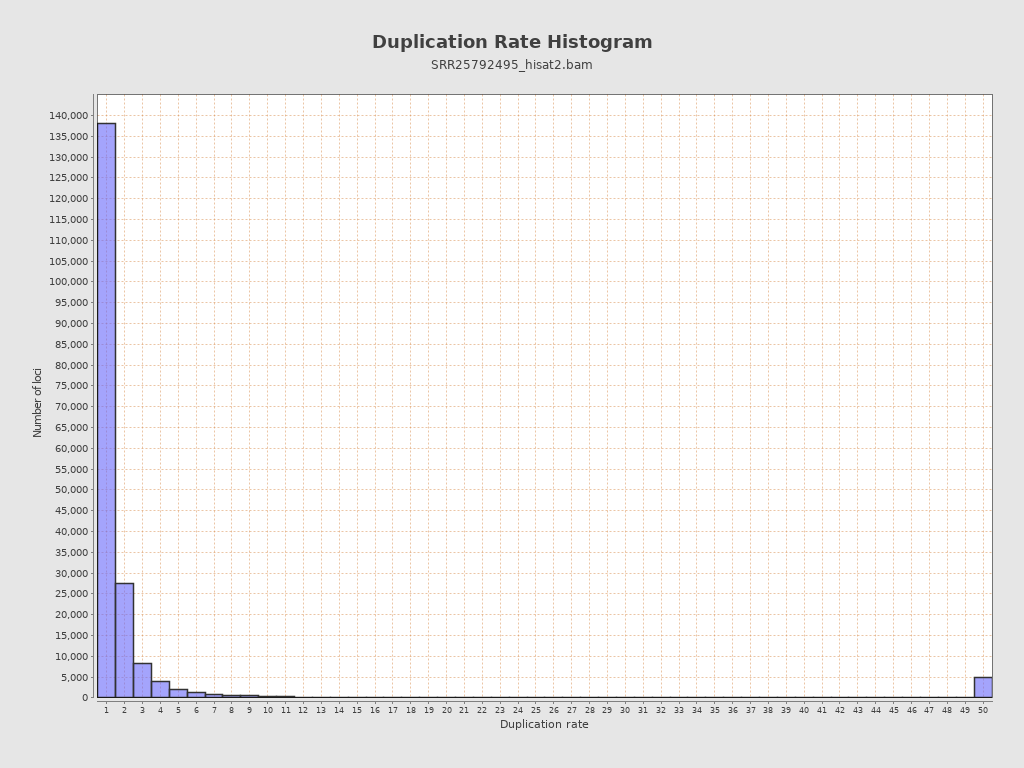
| Duplication rate | 27.61% |
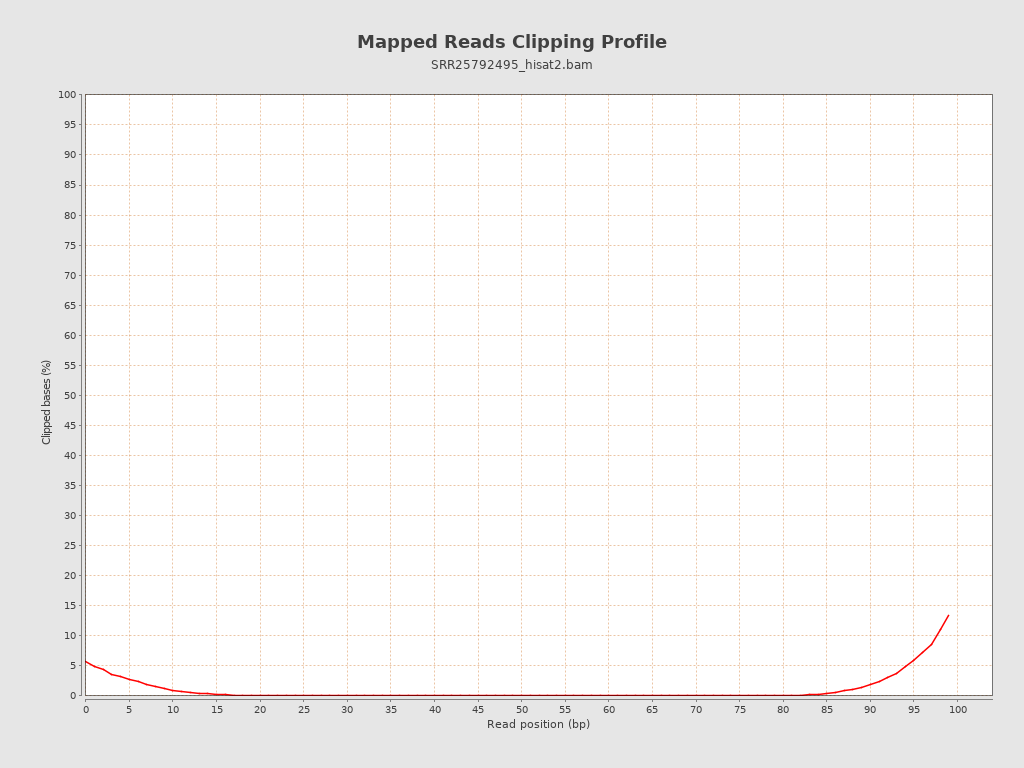
| Clipped reads | 7,161,993 / 20.62% |
ACGT Content
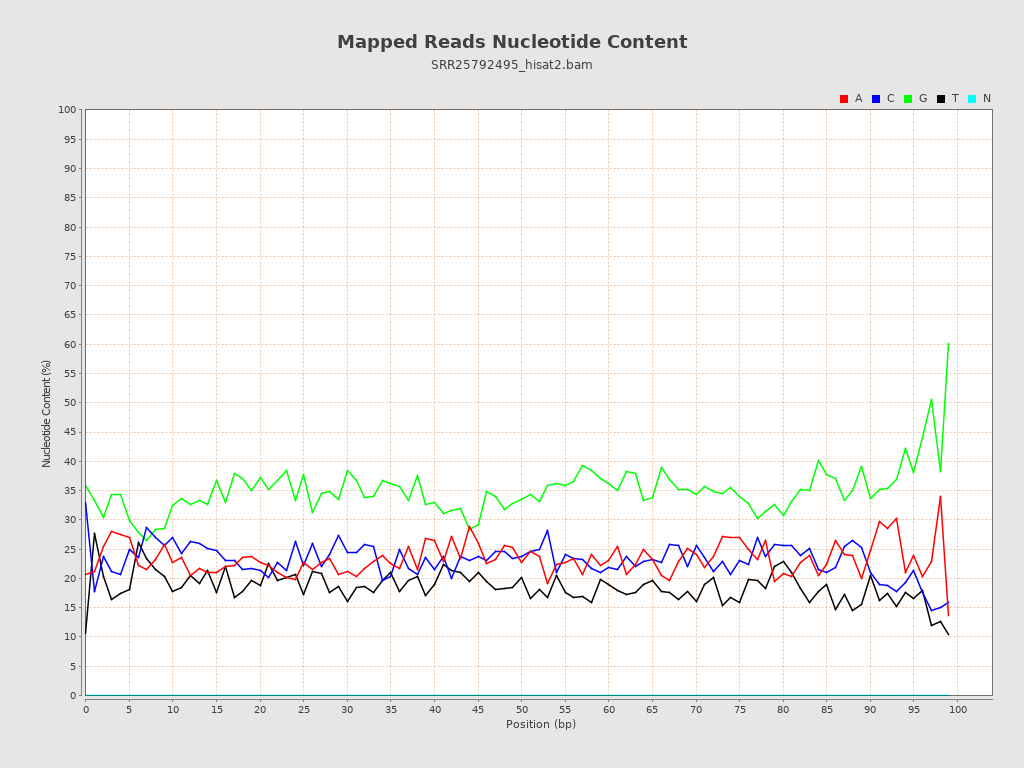
| Number/percentage of A's | 686,445,557 / 23.32% |
| Number/percentage of C's | 680,897,137 / 23.13% |
| Number/percentage of T's | 545,705,997 / 18.54% |
| Number/percentage of G's | 1,030,167,027 / 35% |
| Number/percentage of N's | 14,999,477,074 / 509.63% |
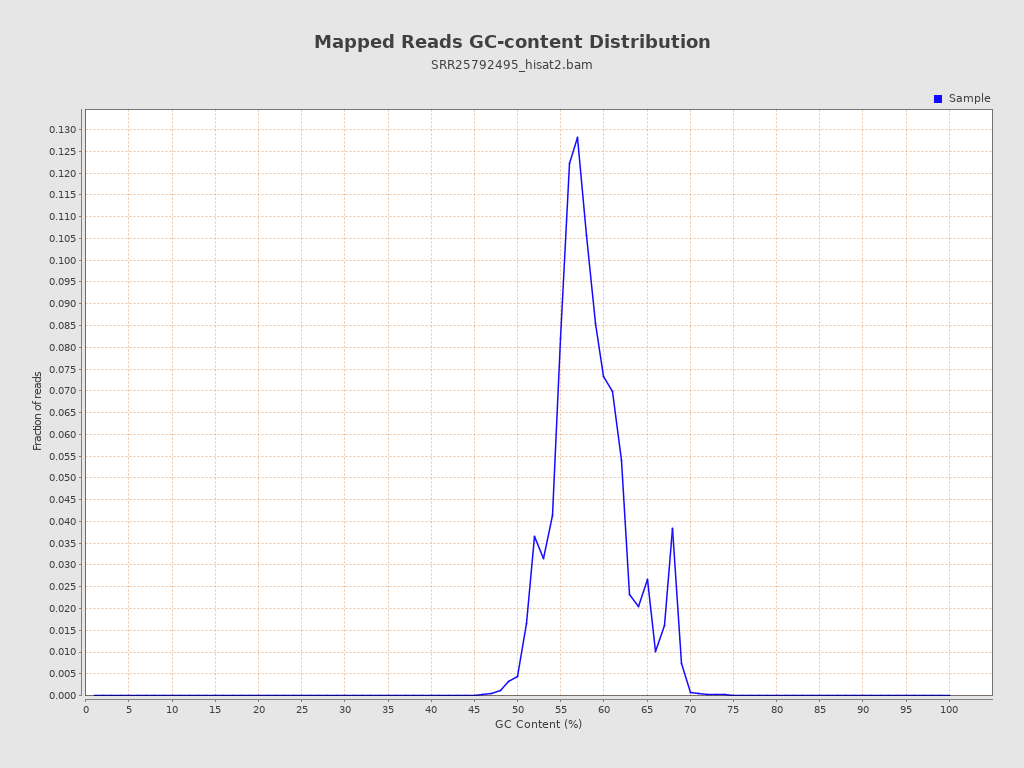
| GC Percentage | 58.14% |
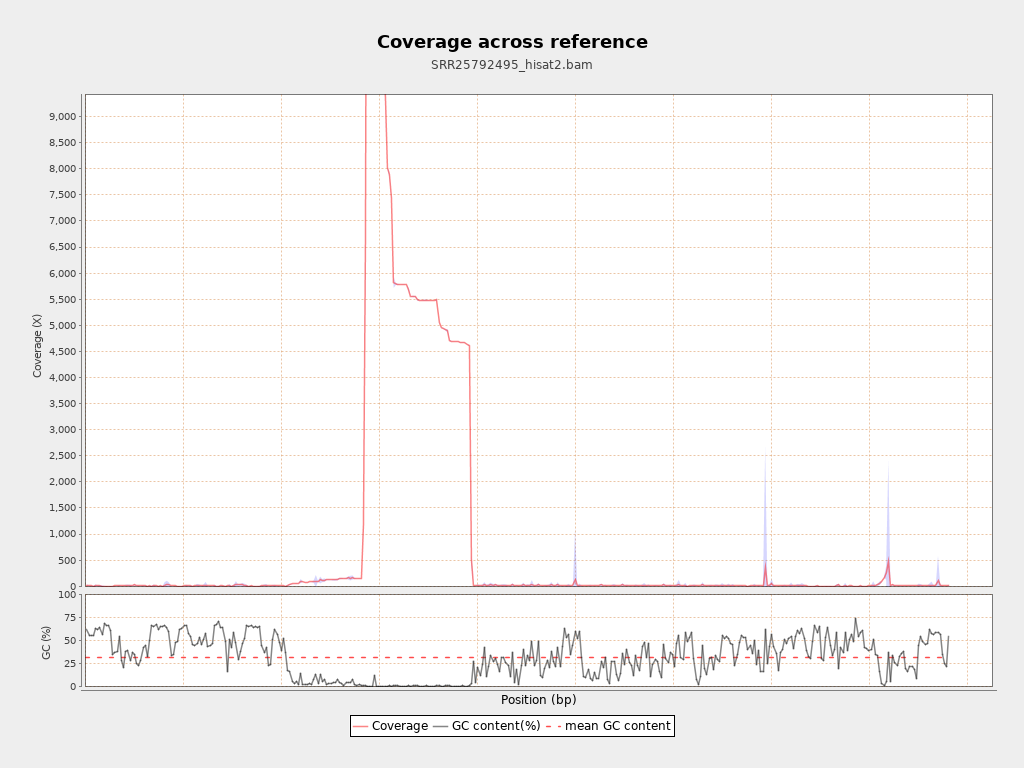
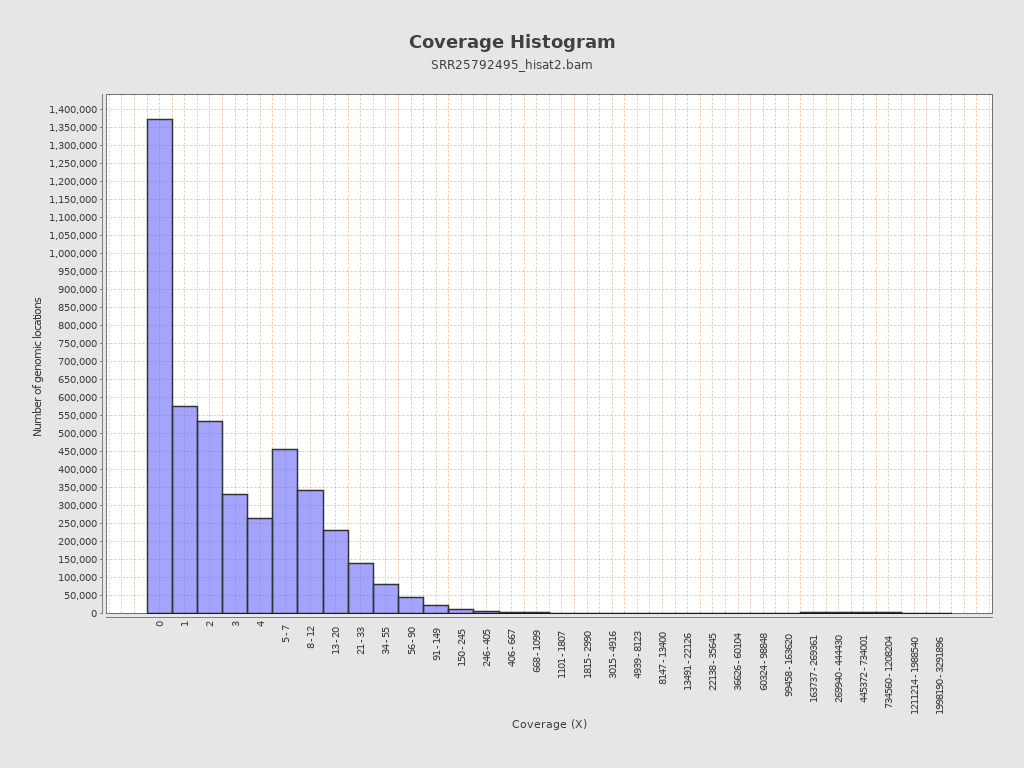
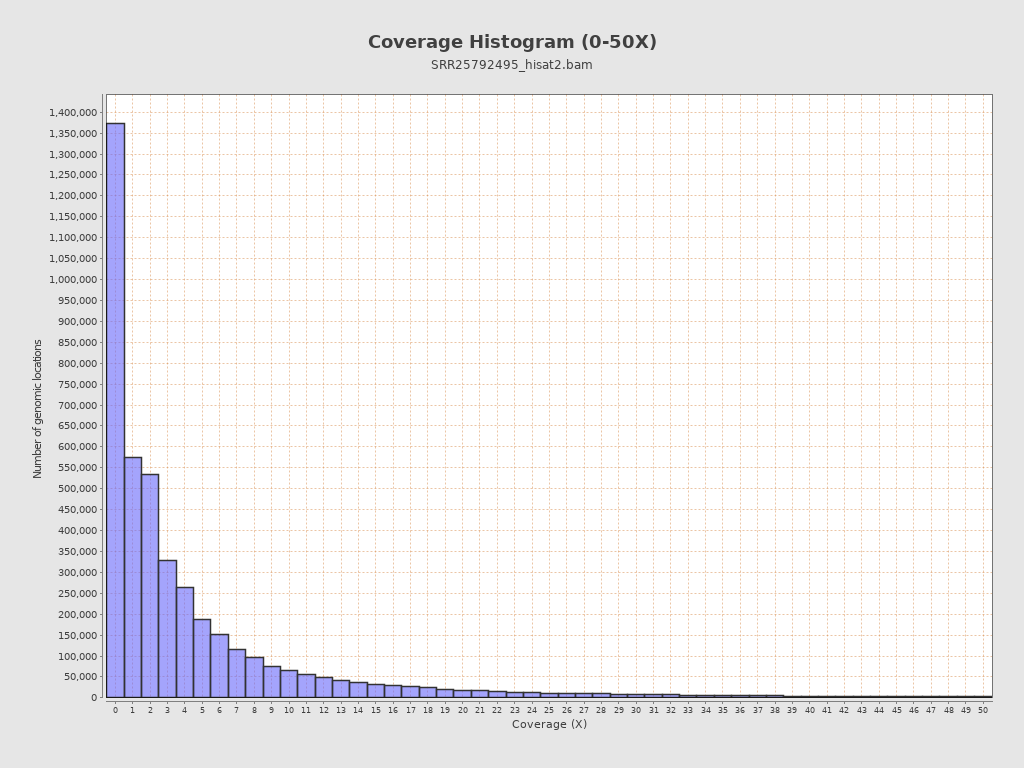
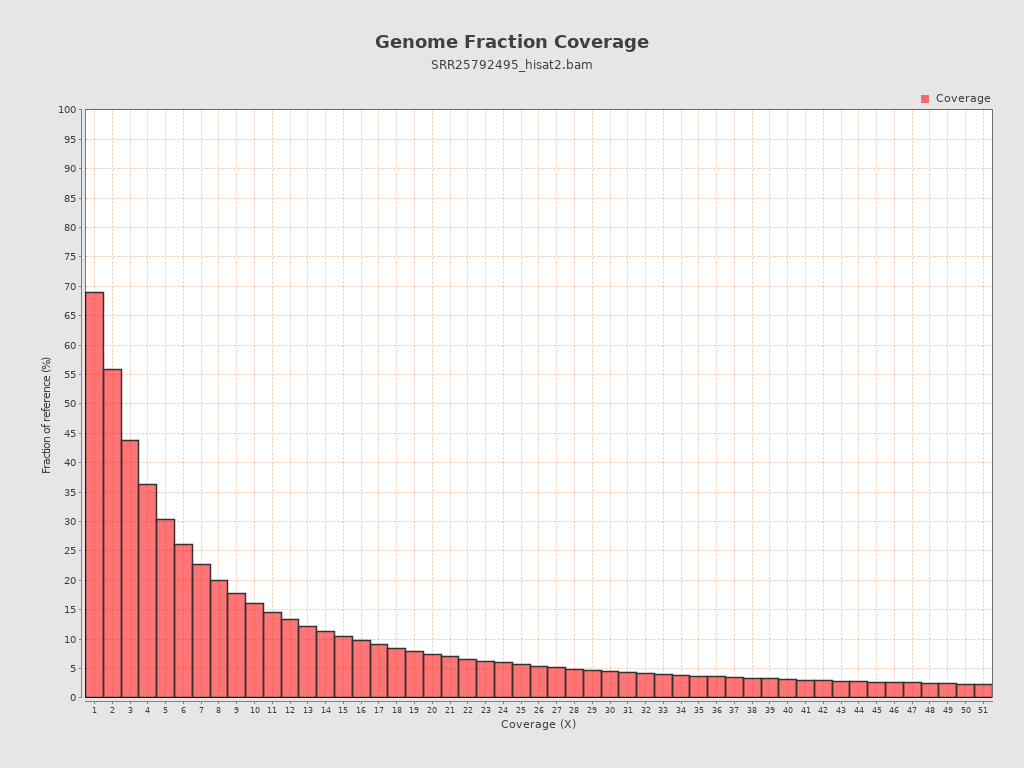
Coverage
| Mean | 4,067.2458 |
| Standard Deviation | 29,124.1095 |
Mapping Quality
| Mean Mapping Quality | 27.78 |
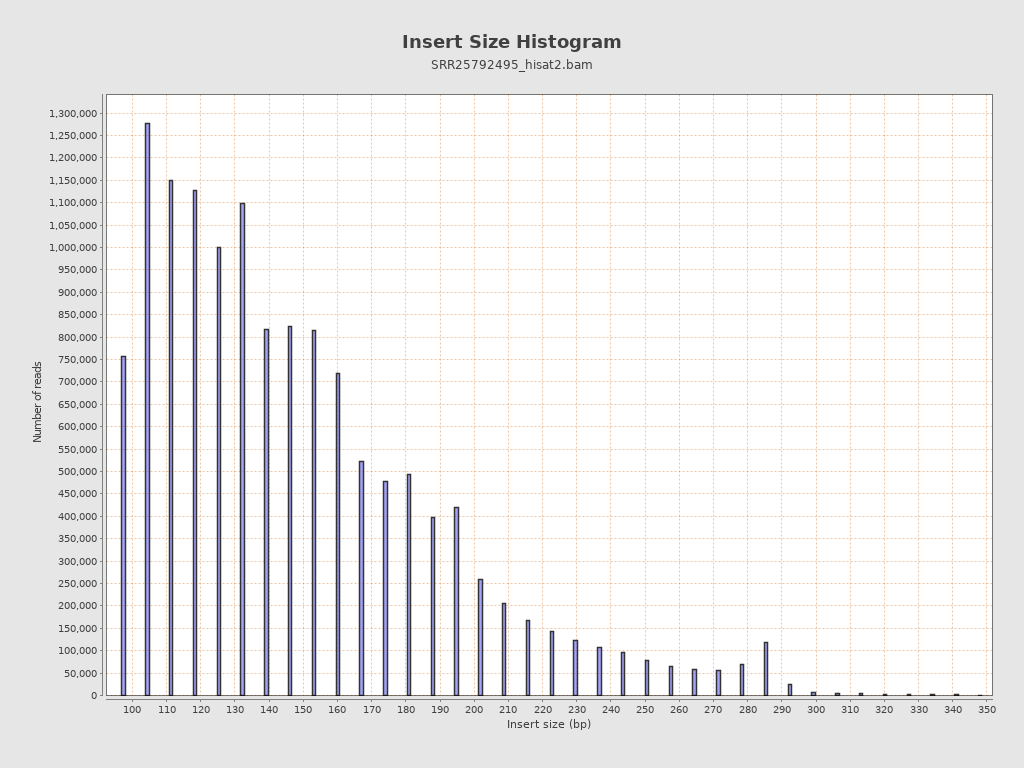
Insert size
| Mean | 280.55 |
| Standard Deviation | 14,783.95 |
| P25/Median/P75 | 120 / 142 / 174 |
Mismatches and indels
| General error rate | 0.03% |
| Mismatches | 4,915,857 |
| Insertions | 5,563 |
| Mapped reads with at least one insertion | 0.02% |
| Deletions | 83,095 |
| Mapped reads with at least one deletion | 0.28% |
| Homopolymer indels | 77.36% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| AL123456.3 | 4411532 | 17942784928 | 4,067.2458 | 29,124.1095 |
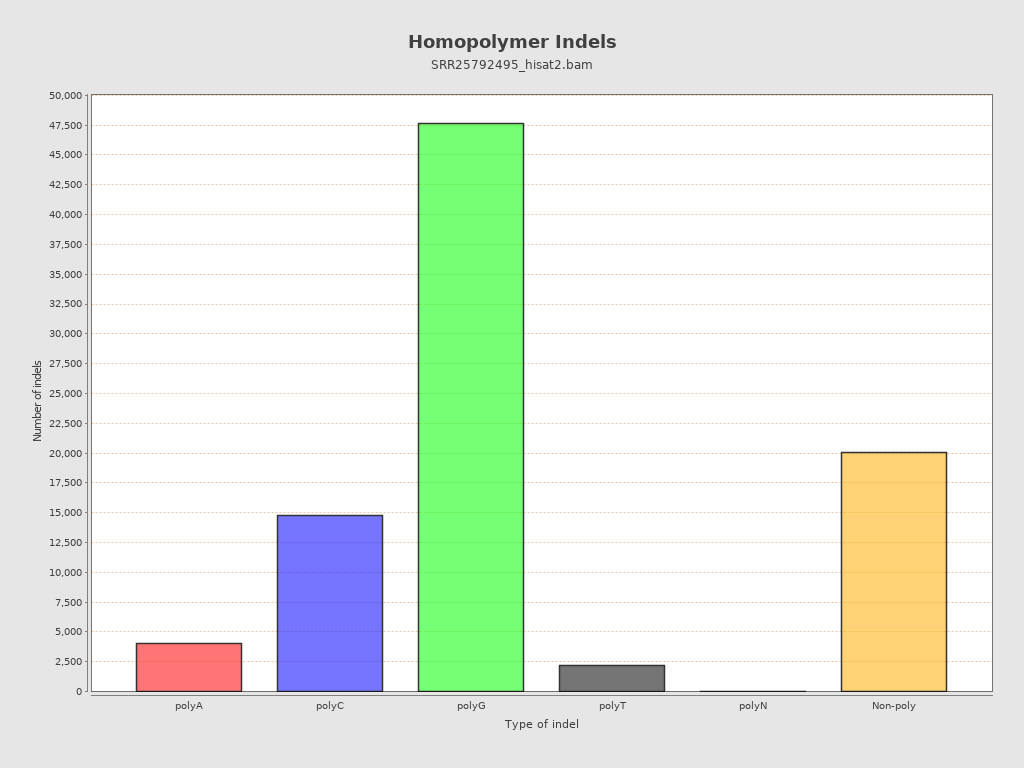
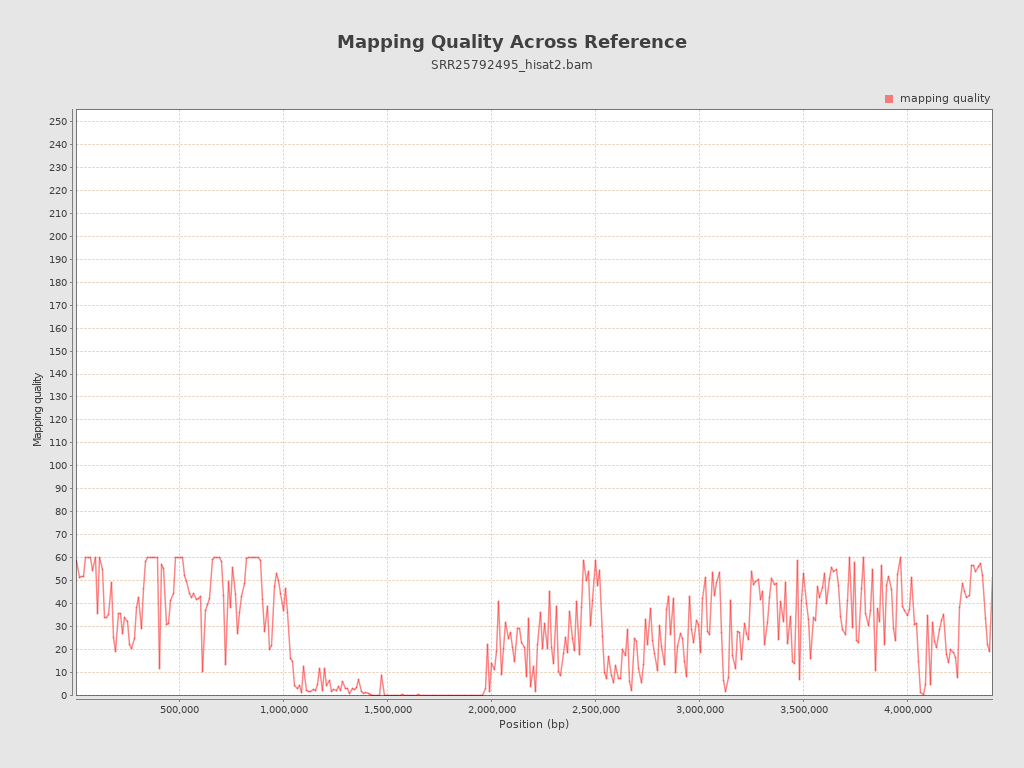
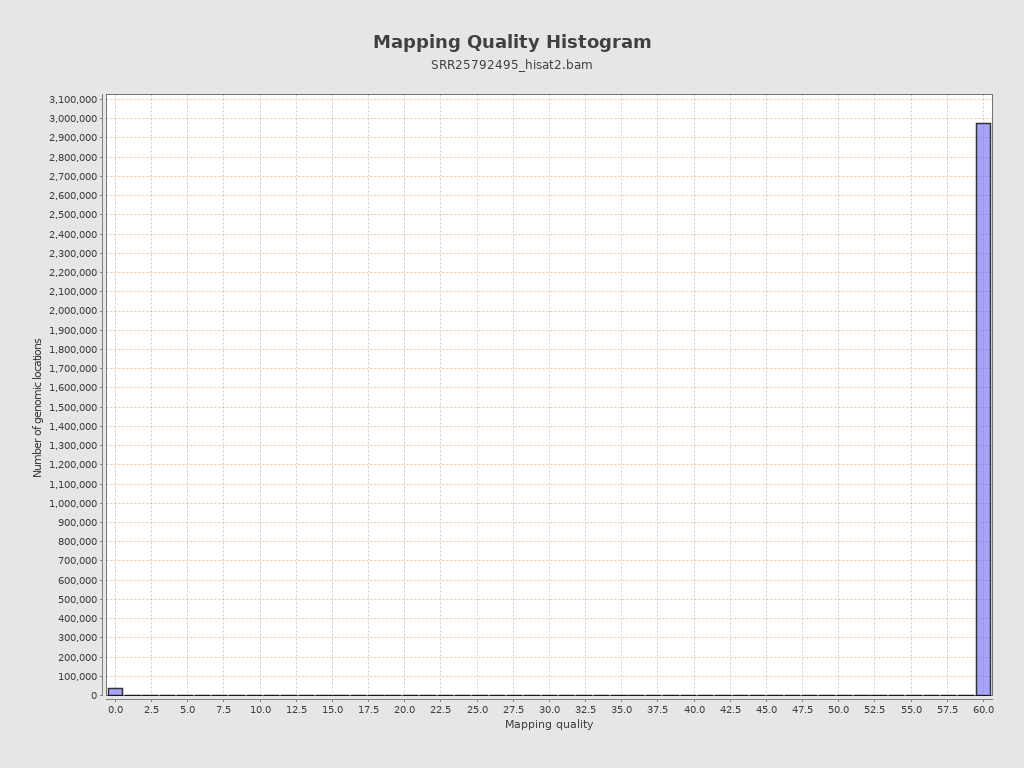
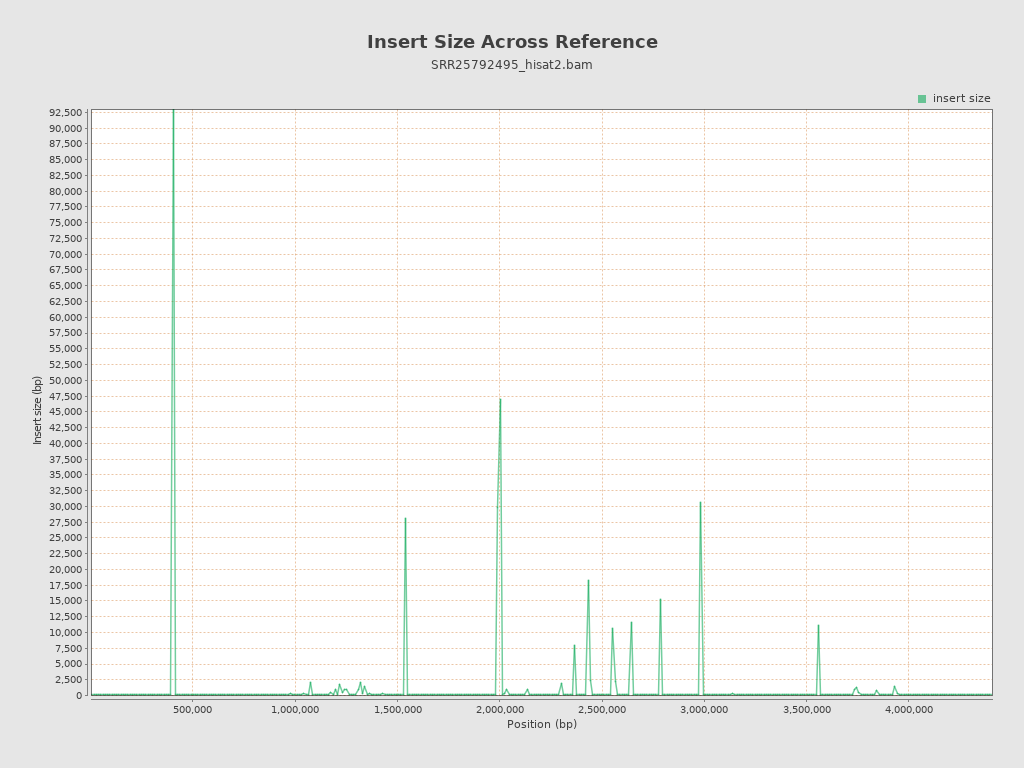
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}