Input data and parameters
QualiMap command line
| qualimap bamqc -bam ../mtuberculosis2/2-alignment/pathogen/bam/mycobacteriumTuberculosis/SRR25792494_hisat2.bam -nw 400 -hm 3 |
Alignment
| Command line: | "/home/dguevara/pipeline/src/../tools/hisat2-2.1.0/hisat2-align-s --wrapper basic-0 -x ../mtuberculosis2/2-alignment/pathogen/indices/hisat2/mycobacteriumTuberculosis -S ../mtuberculosis2/2-alignment/pathogen/sam/mycobacteriumTuberculosis/SRR25792494_hisat2.sam -p 16 -1 ../mtuberculosis2/2-alignment/host/fastq/SRR25792494_1.fastq -2 ../mtuberculosis2/2-alignment/host/fastq/SRR25792494_2.fastq" |
| Draw chromosome limits: | no |
| Analyze overlapping paired-end reads: | no |
| Program: | hisat2 (2.1.0) |
| Analysis date: | Thu Feb 01 04:51:23 CST 2024 |
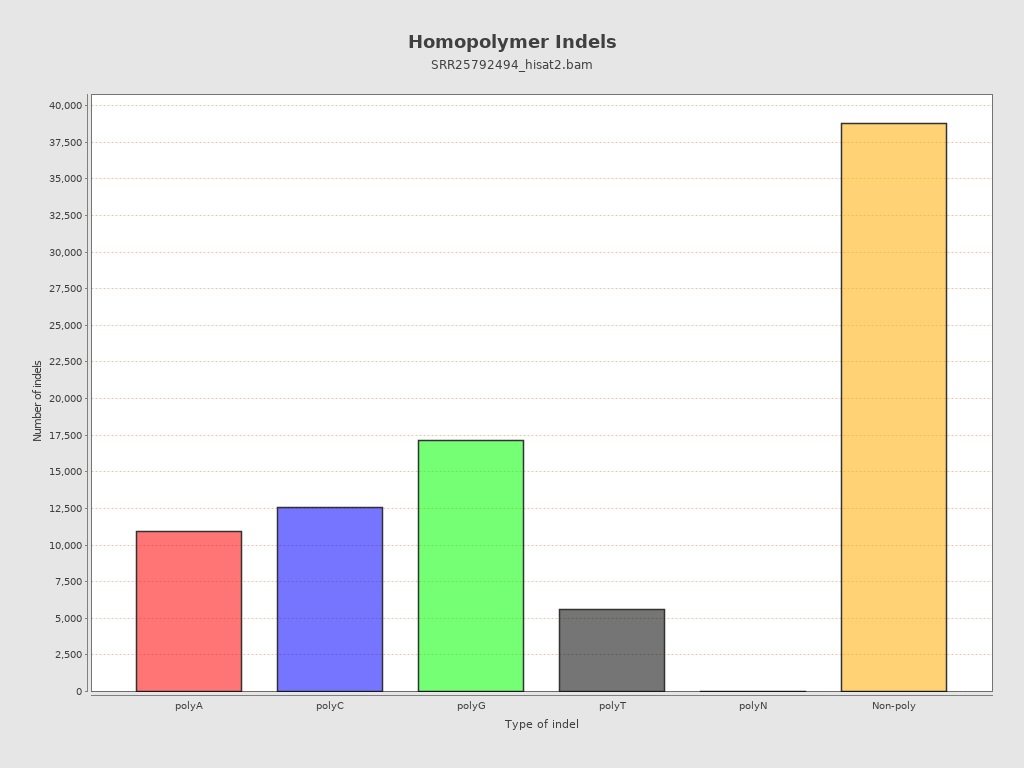
| Size of a homopolymer: | 3 |
| Skip duplicate alignments: | no |
| Number of windows: | 400 |
| BAM file: | ../mtuberculosis2/2-alignment/pathogen/bam/mycobacteriumTuberculosis/SRR25792494_hisat2.bam |
Summary
Globals
| Reference size | 4,411,532 |
| Number of reads | 28,007,727 |
| Mapped reads | 26,959,710 / 96.26% |
| Unmapped reads | 1,048,017 / 3.74% |
| Mapped paired reads | 26,959,710 / 96.26% |
| Mapped reads, first in pair | 13,471,946 / 48.1% |
| Mapped reads, second in pair | 13,487,764 / 48.16% |
| Mapped reads, both in pair | 26,759,367 / 95.54% |
| Mapped reads, singletons | 200,343 / 0.72% |
| Read min/max/mean length | 100 / 100 / 100 |
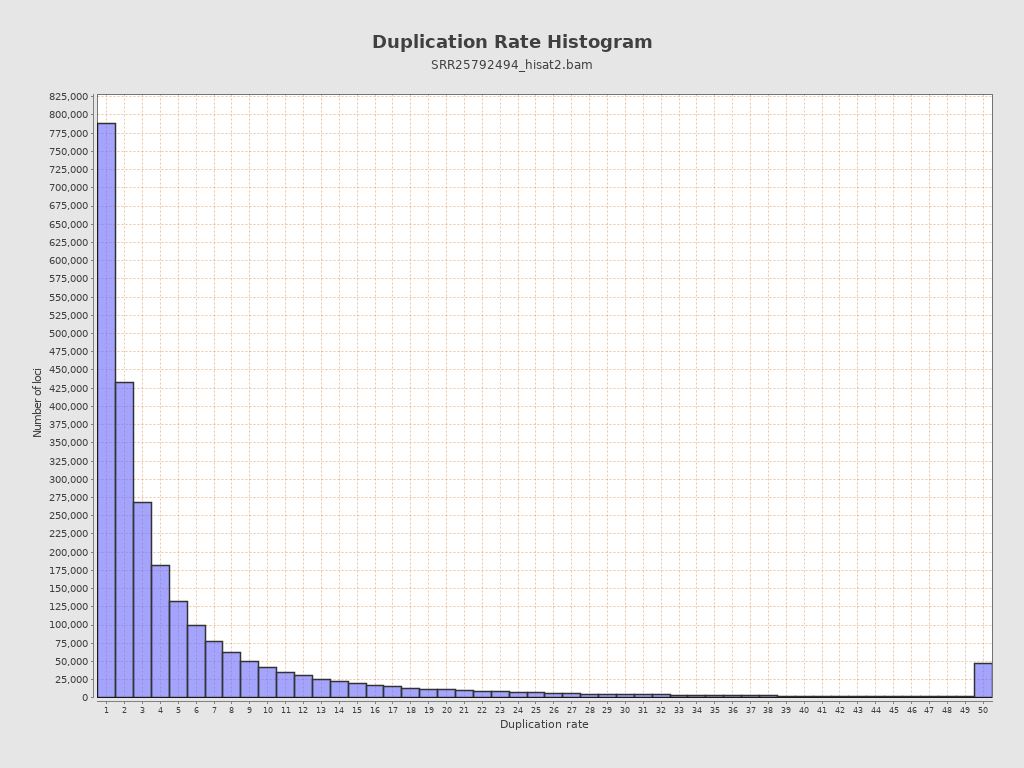
| Duplicated reads (estimated) | 24,463,031 / 87.34% |
| Duplication rate | 68.44% |
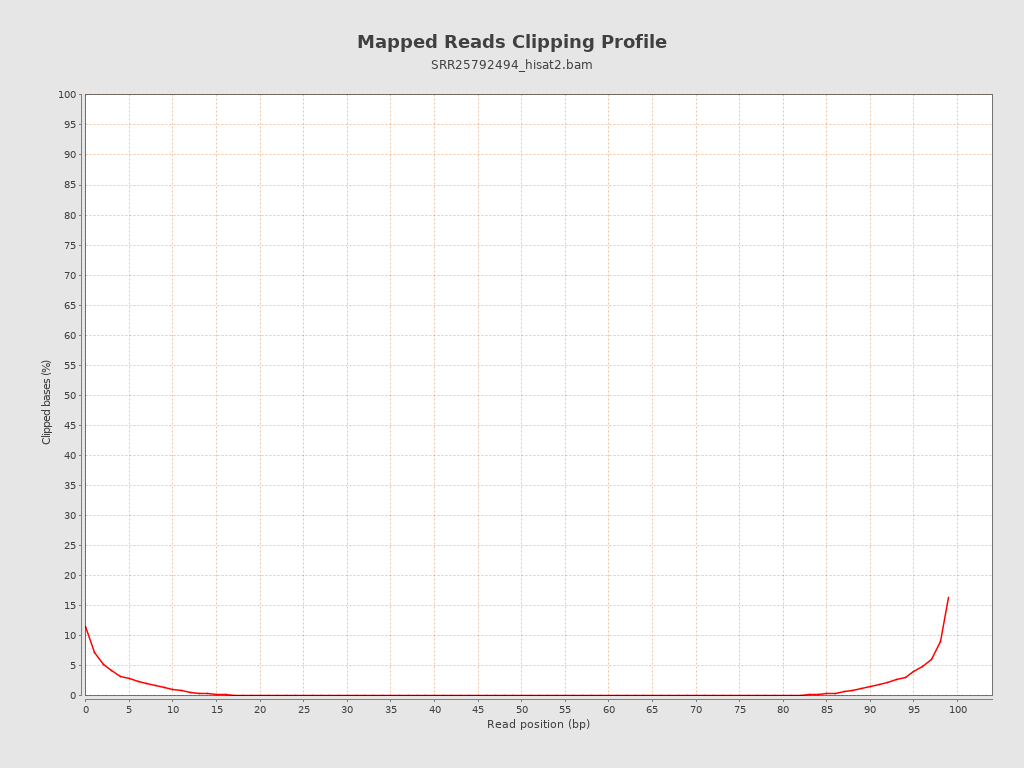
| Clipped reads | 2,300,111 / 8.21% |
ACGT Content
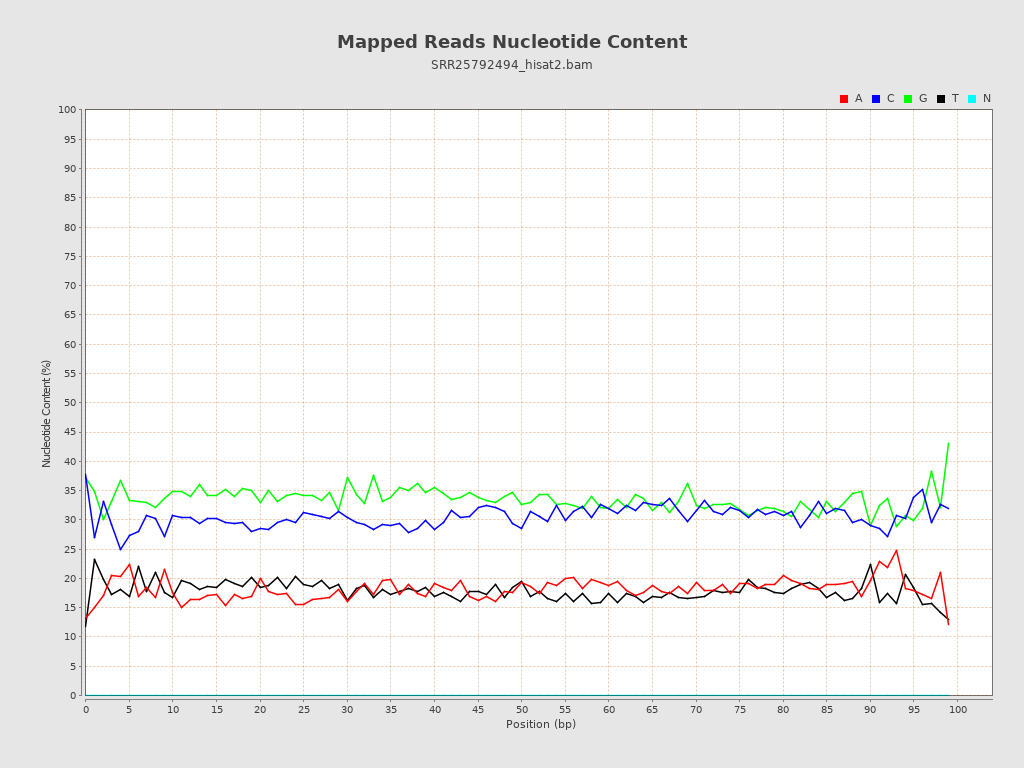
| Number/percentage of A's | 487,939,747 / 18.16% |
| Number/percentage of C's | 821,152,857 / 30.55% |
| Number/percentage of T's | 478,401,027 / 17.8% |
| Number/percentage of G's | 899,987,629 / 33.49% |
| Number/percentage of N's | 165,177,243,264 / 6146.17% |
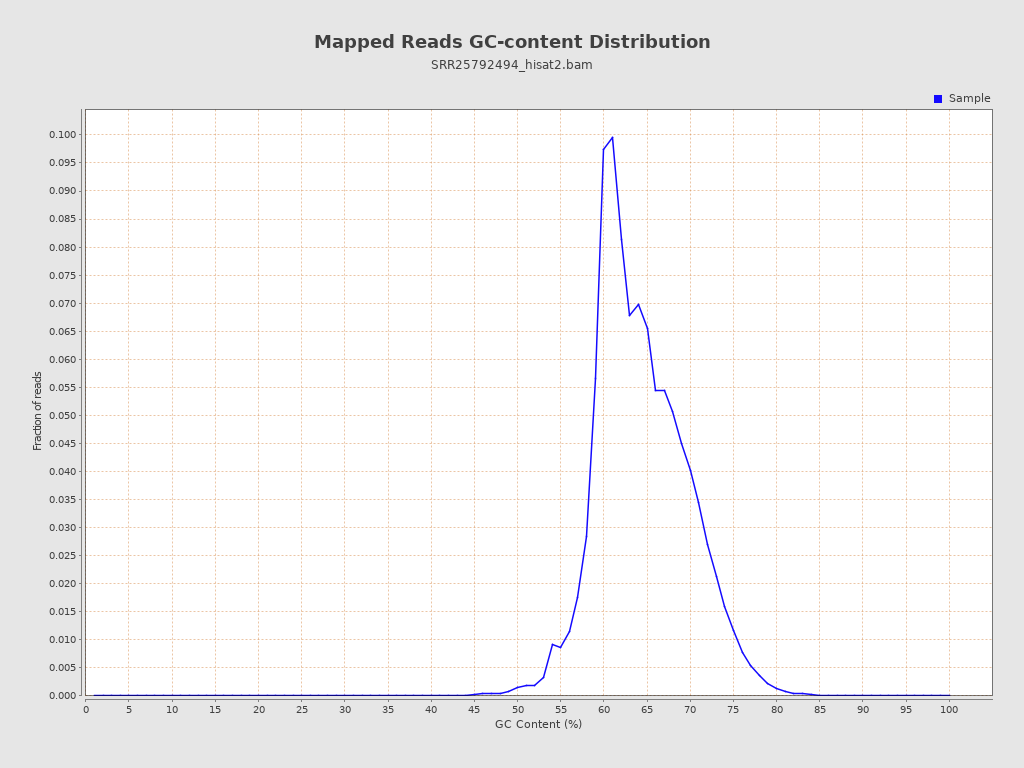
| GC Percentage | 64.04% |
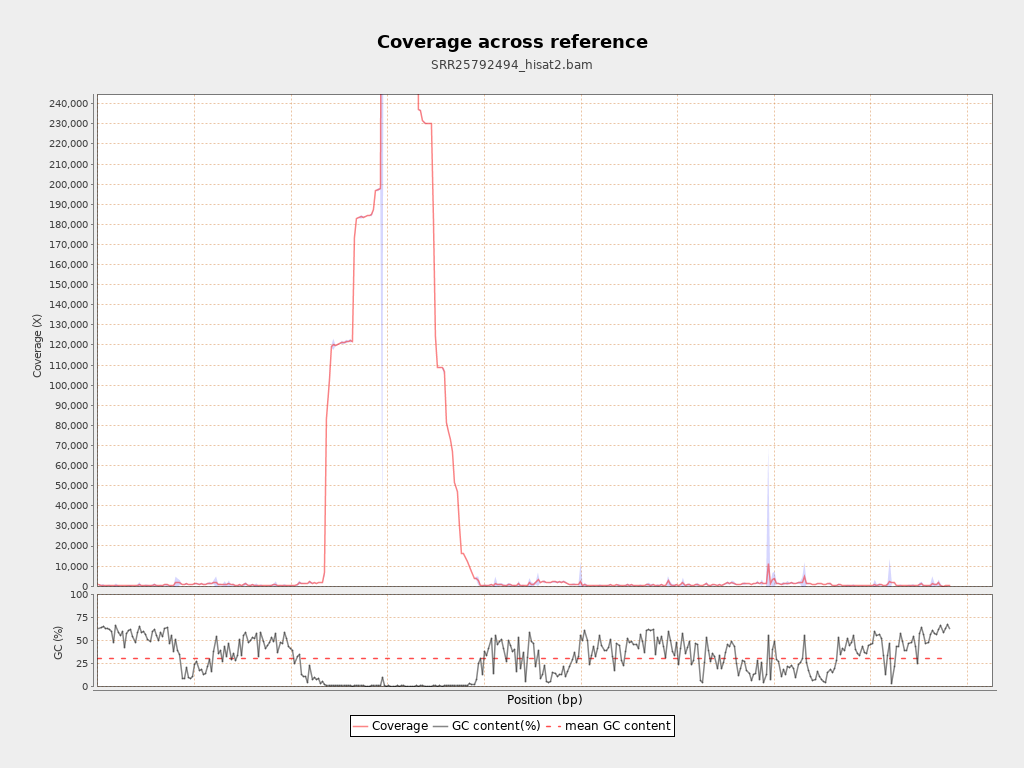
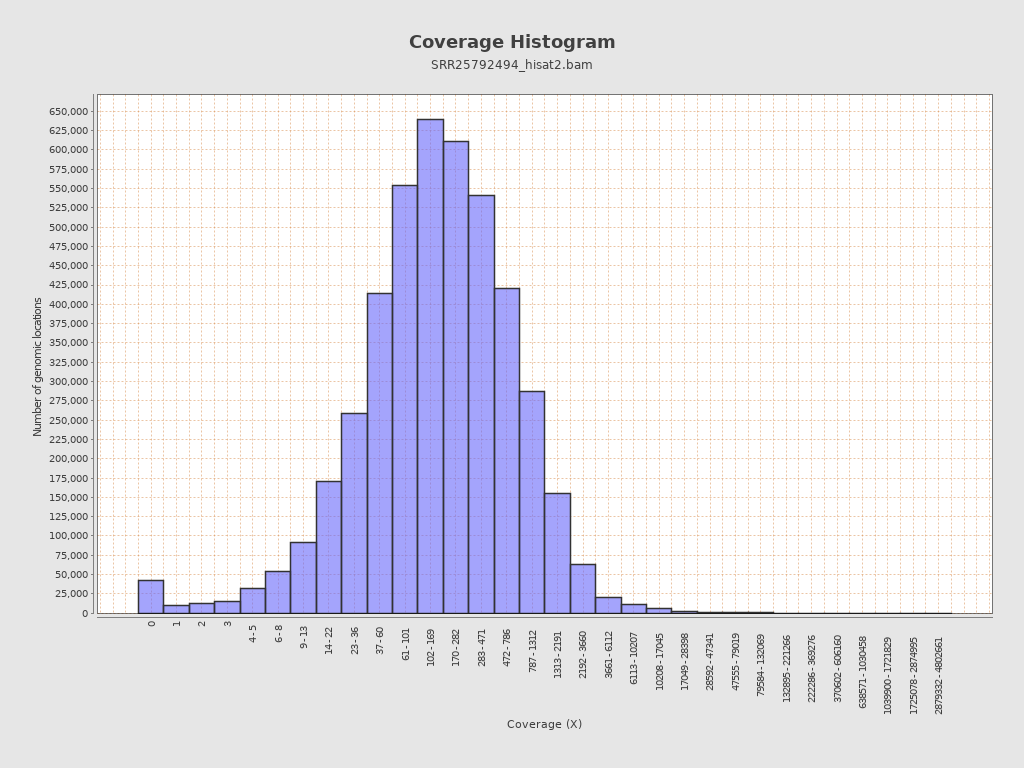
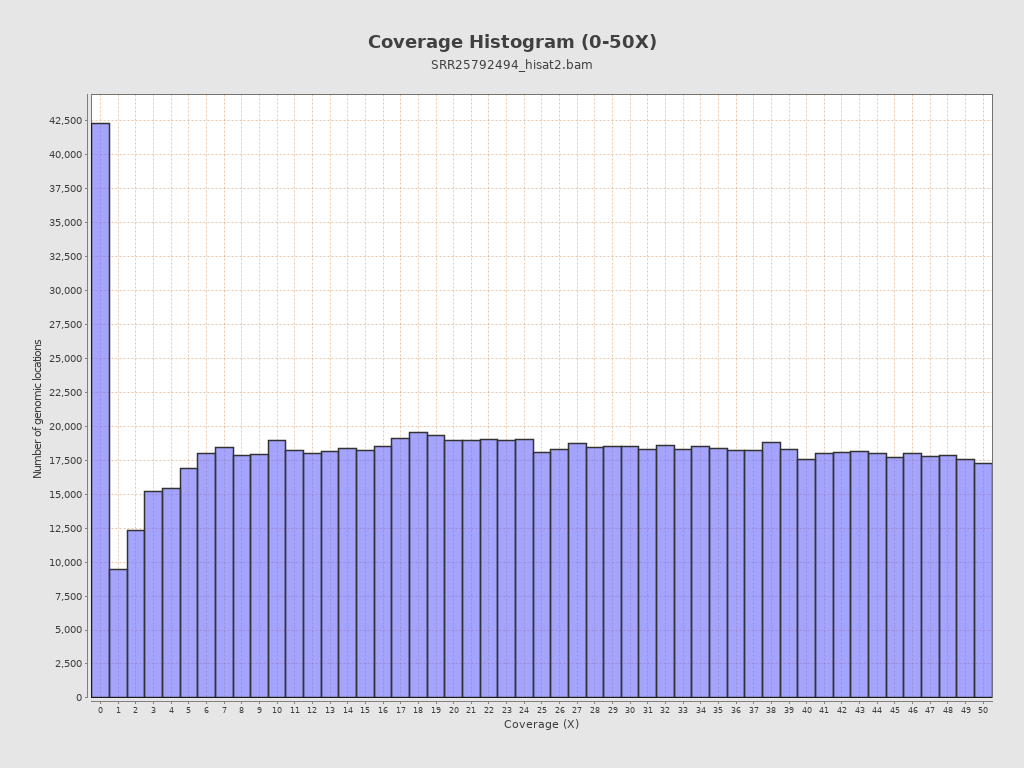
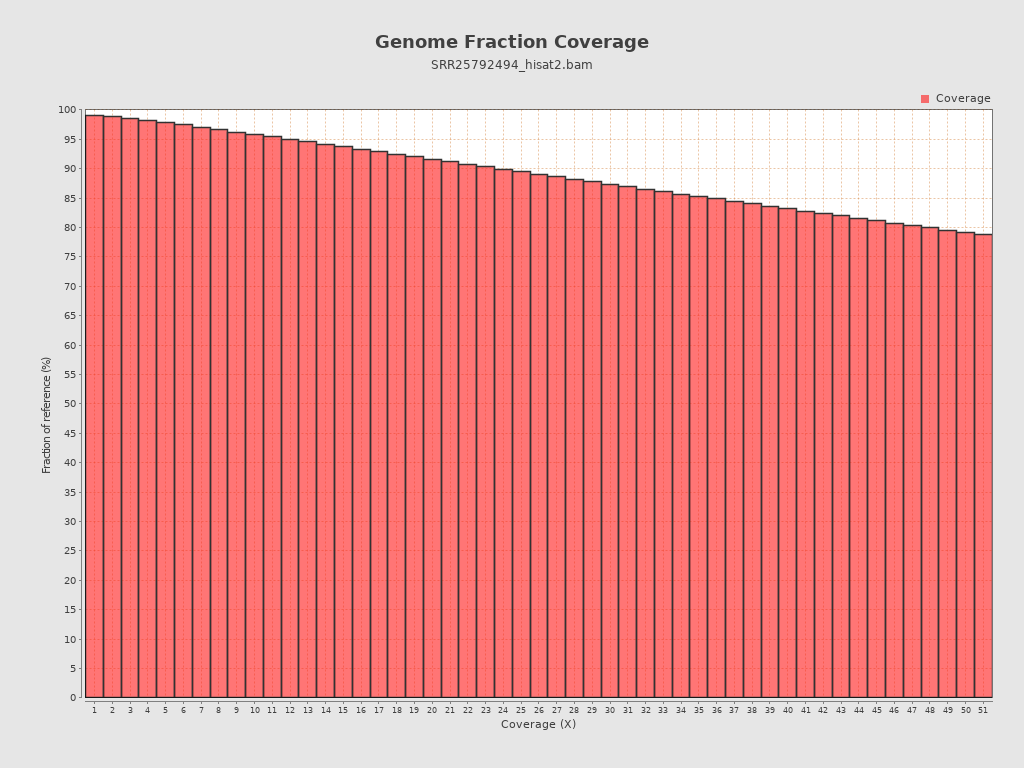
Coverage
| Mean | 38,051.3684 |
| Standard Deviation | 43,773.384 |
Mapping Quality
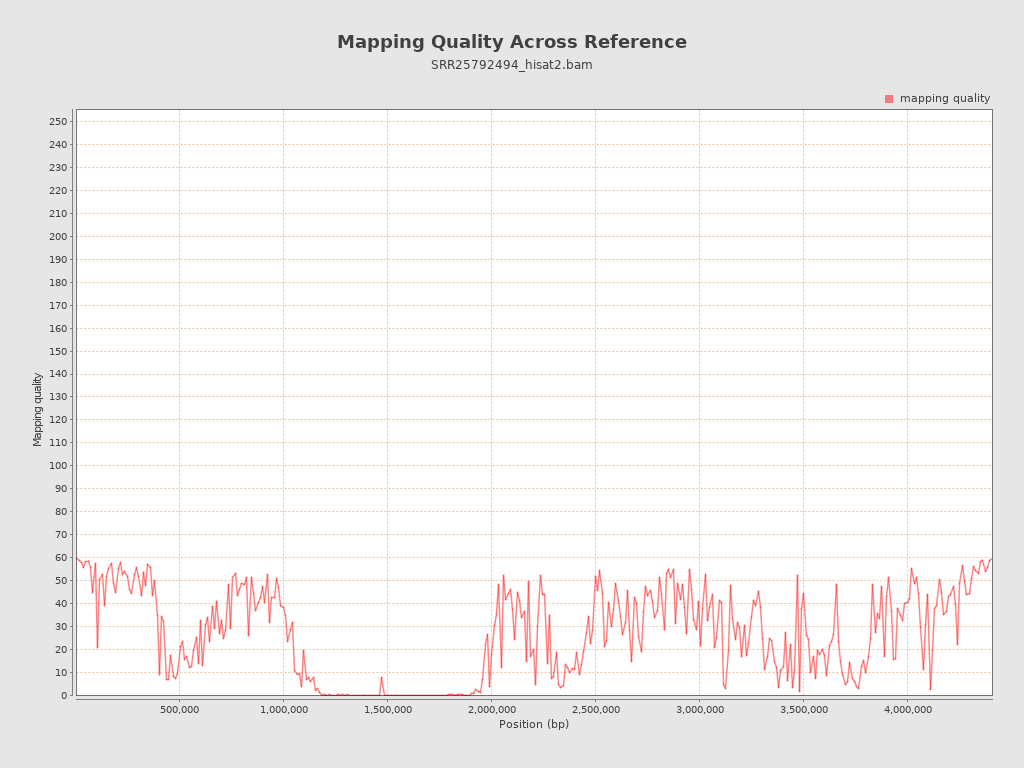
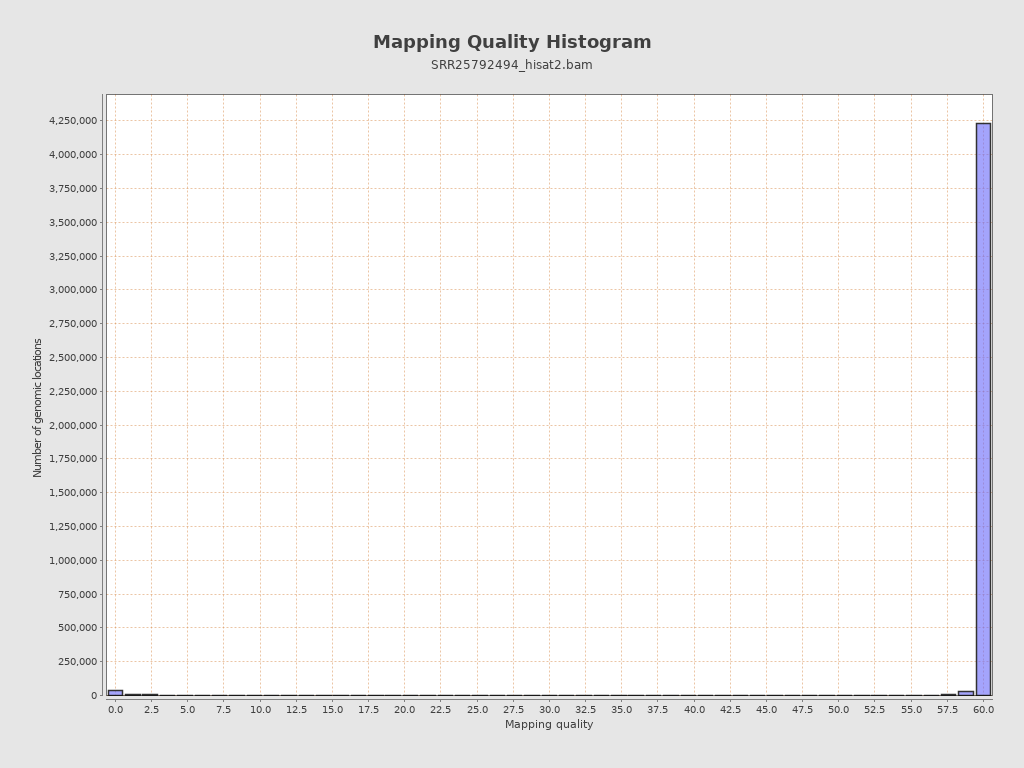
| Mean Mapping Quality | 26.72 |
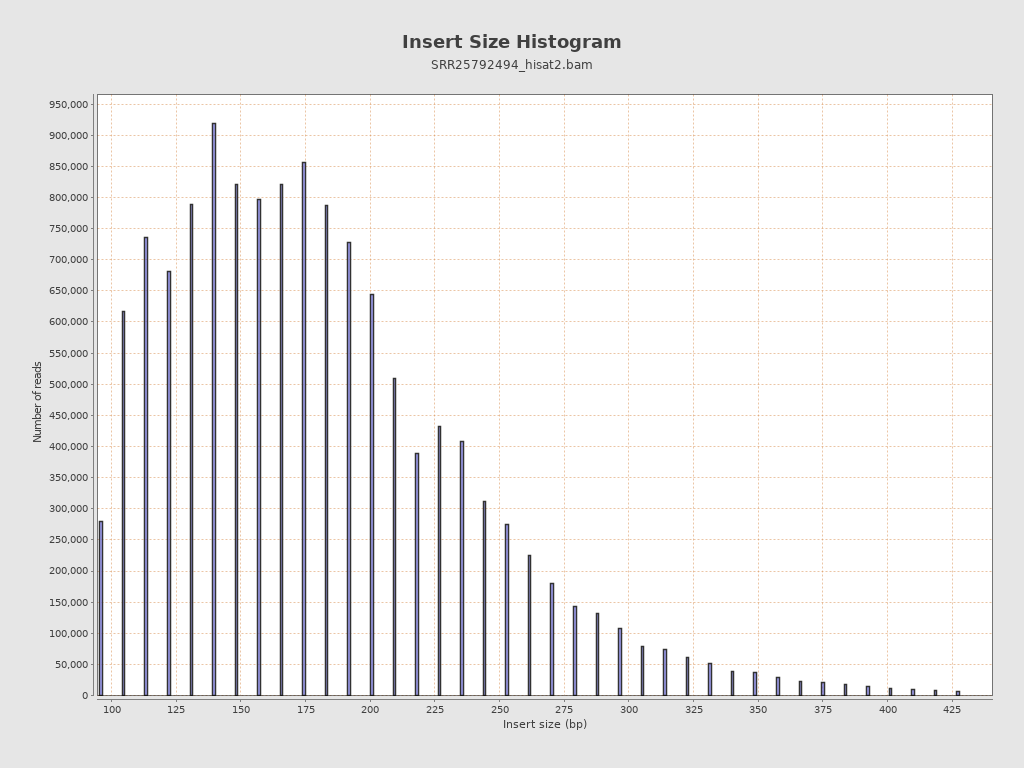
Insert size
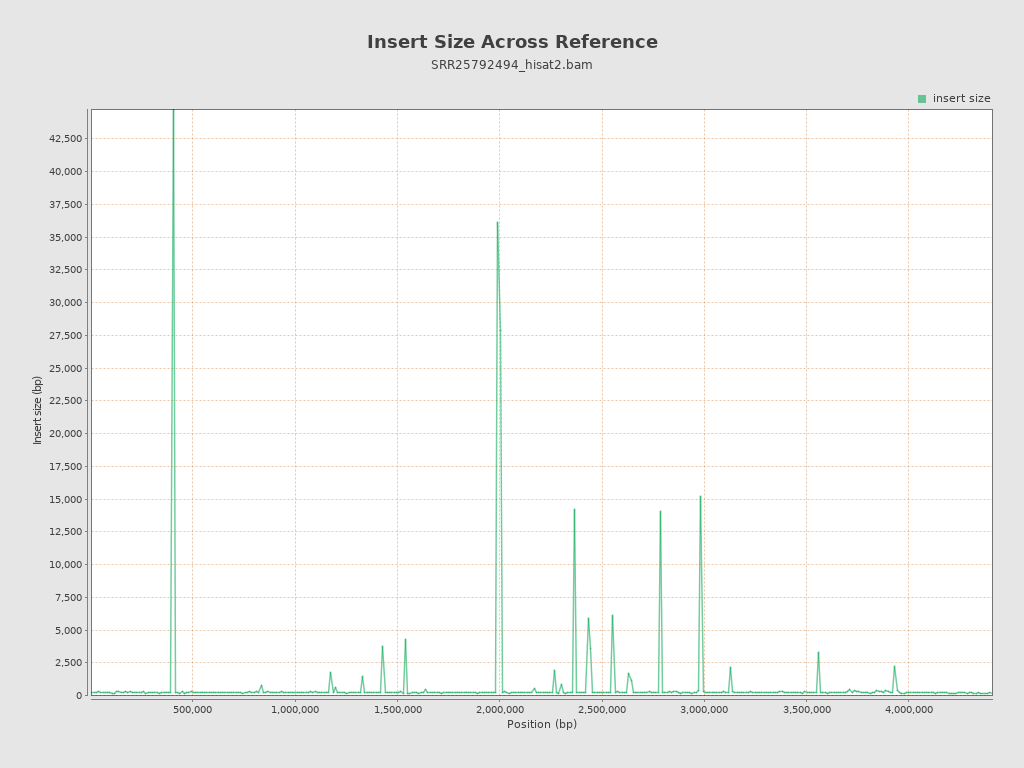
| Mean | 7,005.67 |
| Standard Deviation | 114,258.56 |
| P25/Median/P75 | 142 / 176 / 218 |
Mismatches and indels
| General error rate | 0% |
| Mismatches | 3,575,158 |
| Insertions | 11,232 |
| Mapped reads with at least one insertion | 0.04% |
| Deletions | 73,750 |
| Mapped reads with at least one deletion | 0.27% |
| Homopolymer indels | 54.35% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| AL123456.3 | 4411532 | 167864829252 | 38,051.3684 | 43,773.384 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}