Input data and parameters
QualiMap command line
| qualimap bamqc -bam ../mtuberculosis2/2-alignment/pathogen/bam/mycobacteriumTuberculosis/SRR25787975_hisat2.bam -nw 400 -hm 3 |
Alignment
| Command line: | "/home/dguevara/pipeline/src/../tools/hisat2-2.1.0/hisat2-align-s --wrapper basic-0 -x ../mtuberculosis2/2-alignment/pathogen/indices/hisat2/mycobacteriumTuberculosis -S ../mtuberculosis2/2-alignment/pathogen/sam/mycobacteriumTuberculosis/SRR25787975_hisat2.sam -p 16 -1 ../mtuberculosis2/2-alignment/host/fastq/SRR25787975_1.fastq -2 ../mtuberculosis2/2-alignment/host/fastq/SRR25787975_2.fastq" |
| Draw chromosome limits: | no |
| Analyze overlapping paired-end reads: | no |
| Program: | hisat2 (2.1.0) |
| Analysis date: | Thu Feb 01 04:33:55 CST 2024 |
| Size of a homopolymer: | 3 |
| Skip duplicate alignments: | no |
| Number of windows: | 400 |
| BAM file: | ../mtuberculosis2/2-alignment/pathogen/bam/mycobacteriumTuberculosis/SRR25787975_hisat2.bam |
Summary
Globals
| Reference size | 4,411,532 |
| Number of reads | 32,347,422 |
| Mapped reads | 30,540,696 / 94.41% |
| Unmapped reads | 1,806,726 / 5.59% |
| Mapped paired reads | 30,540,696 / 94.41% |
| Mapped reads, first in pair | 15,053,461 / 46.54% |
| Mapped reads, second in pair | 15,487,235 / 47.88% |
| Mapped reads, both in pair | 30,179,767 / 93.3% |
| Mapped reads, singletons | 360,929 / 1.12% |
| Read min/max/mean length | 100 / 100 / 100 |
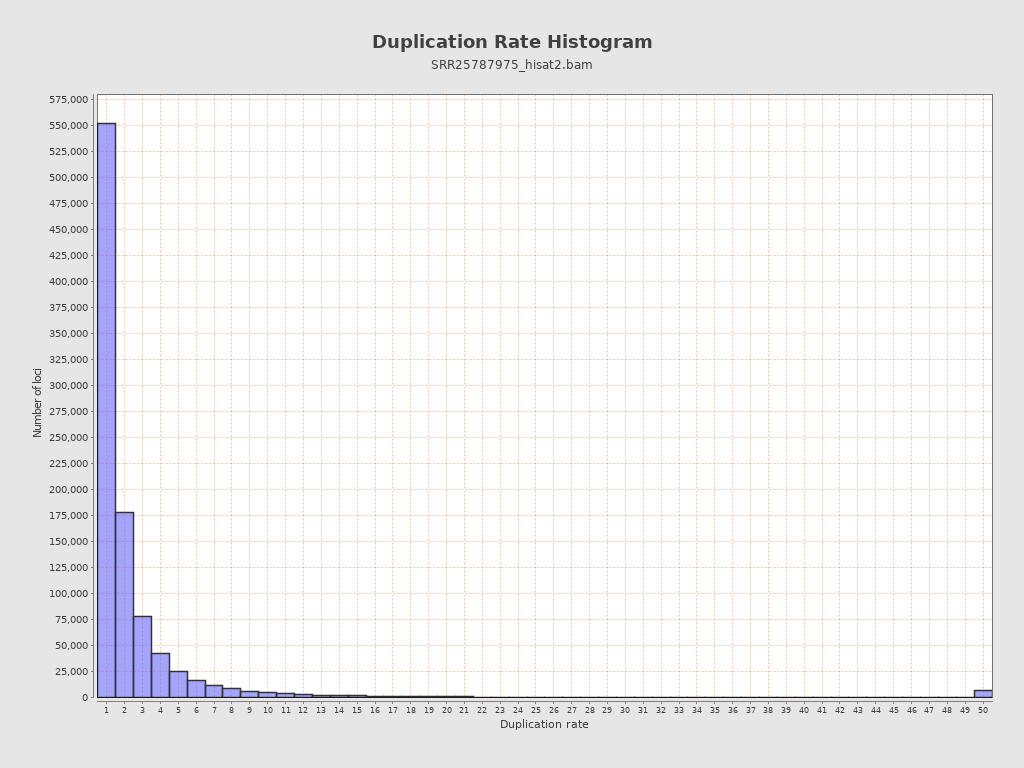
| Duplicated reads (estimated) | 29,579,016 / 91.44% |
| Duplication rate | 42.6% |
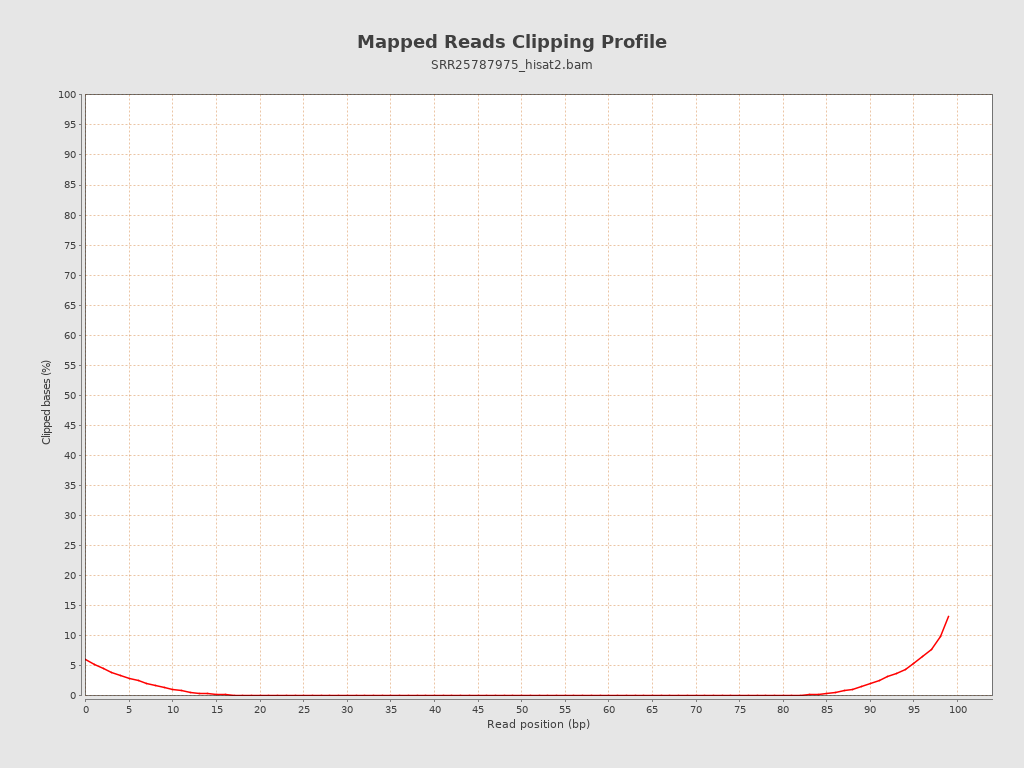
| Clipped reads | 4,657,765 / 14.4% |
ACGT Content
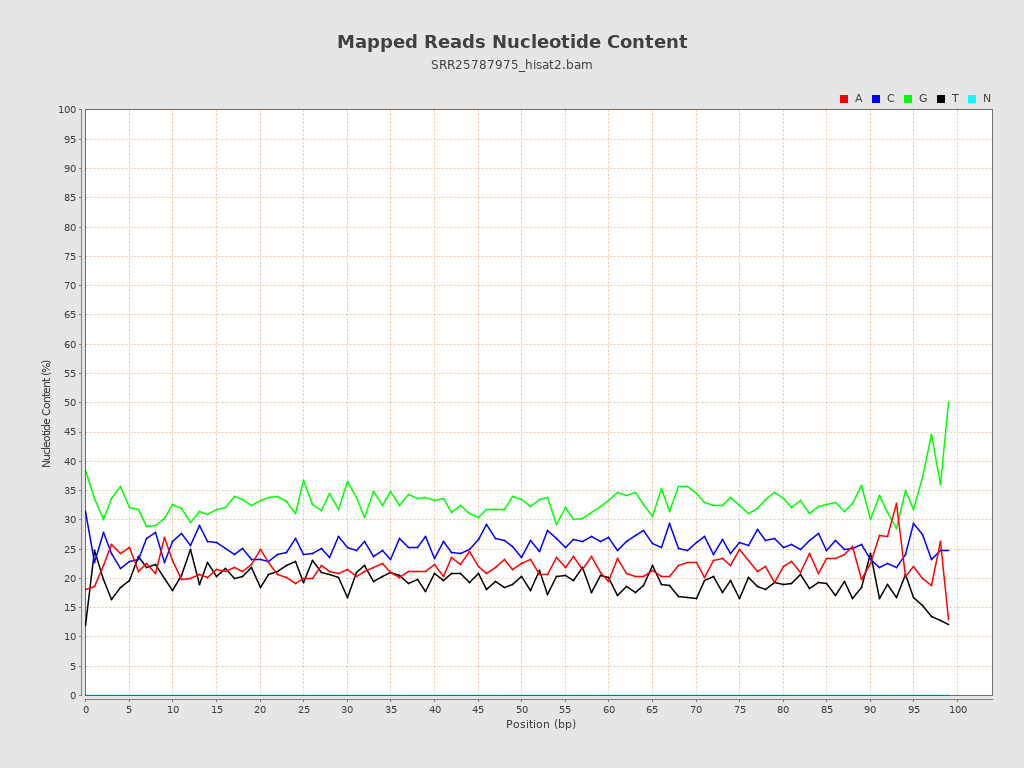
| Number/percentage of A's | 665,947,440 / 21.99% |
| Number/percentage of C's | 774,121,281 / 25.56% |
| Number/percentage of T's | 587,669,493 / 19.4% |
| Number/percentage of G's | 1,001,181,656 / 33.05% |
| Number/percentage of N's | 53,193,601,872 / 1756.19% |
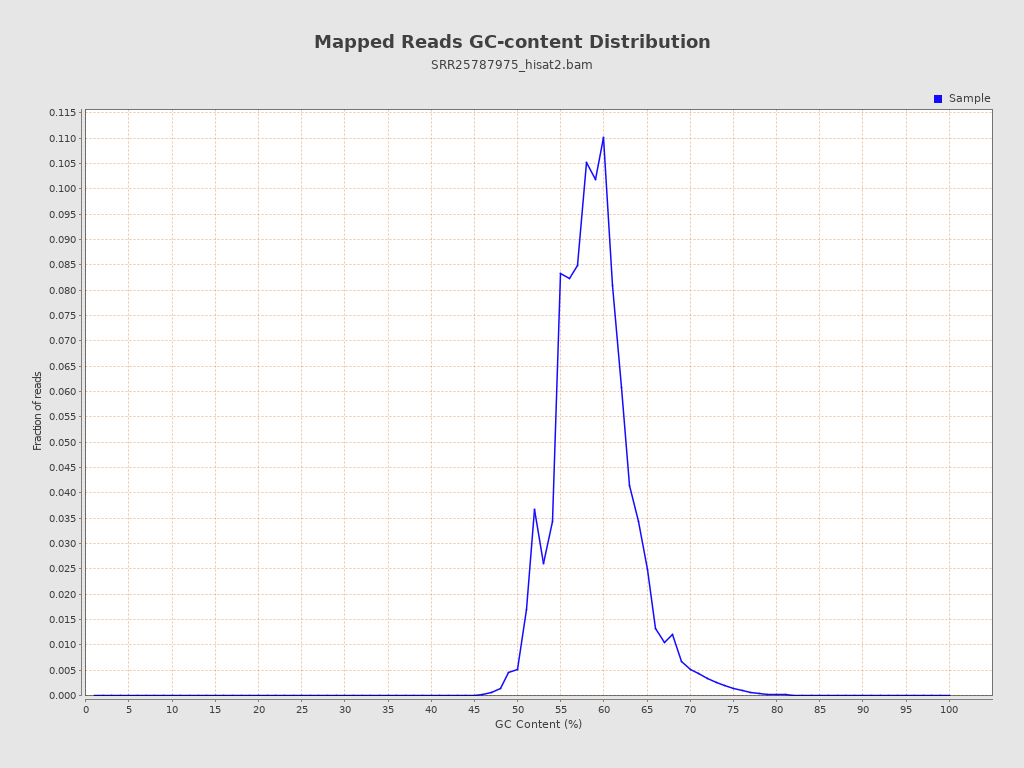
| GC Percentage | 58.61% |
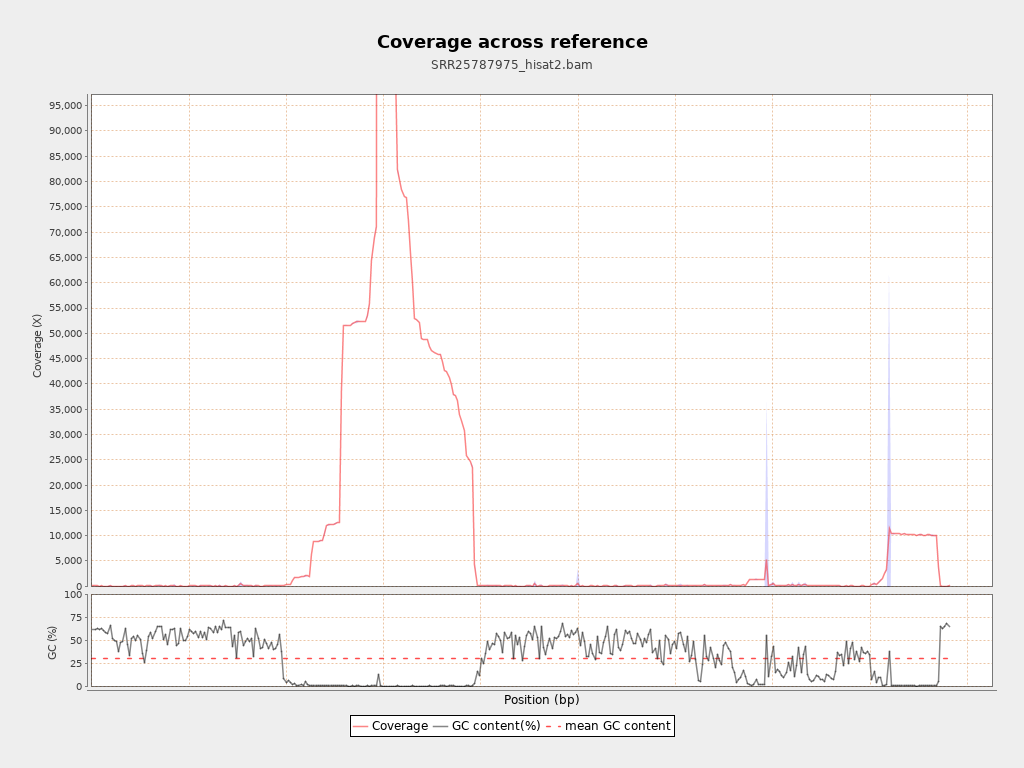
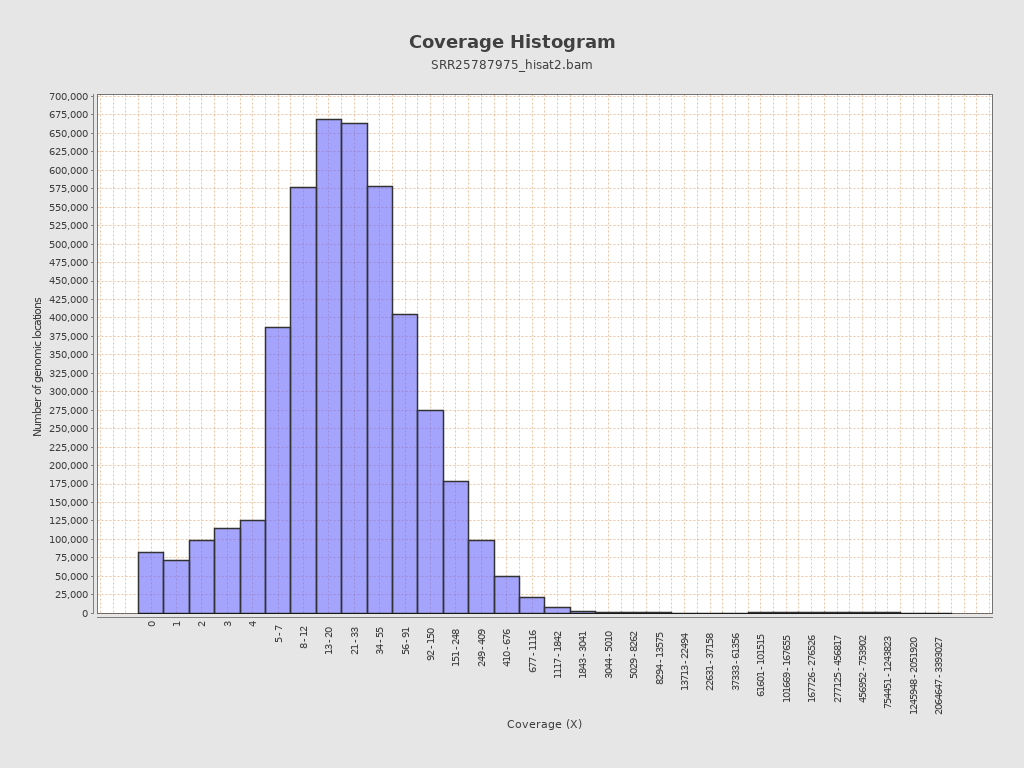
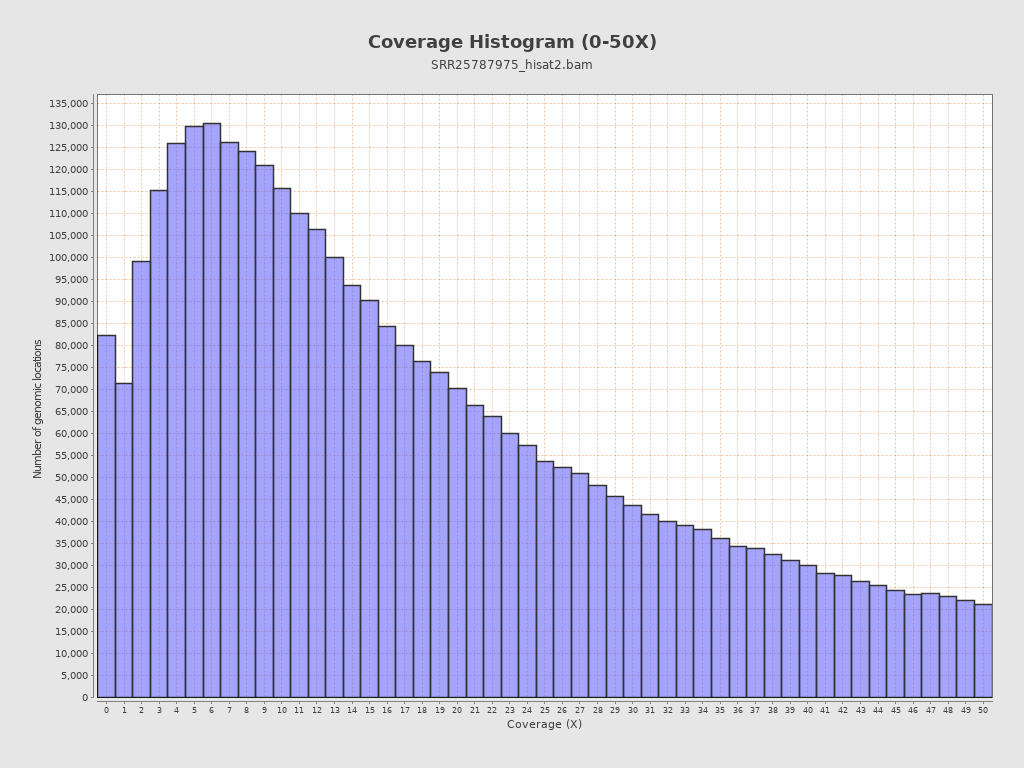
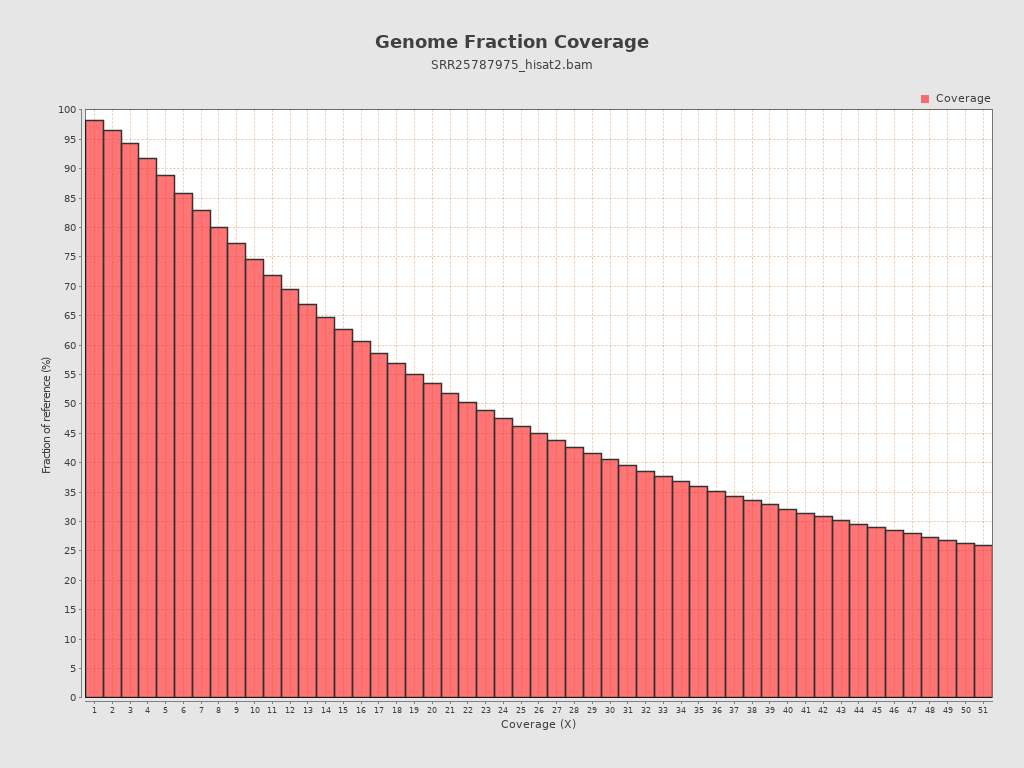
Coverage
| Mean | 12,744.4616 |
| Standard Deviation | 29,737.1205 |
Mapping Quality
| Mean Mapping Quality | 26.12 |
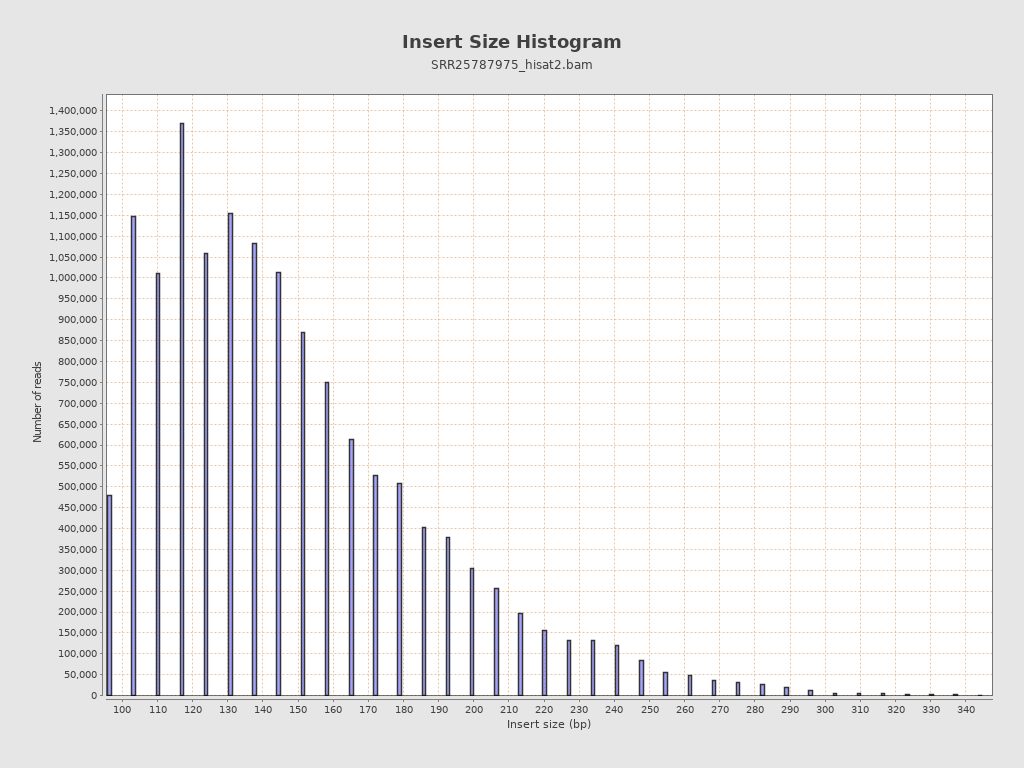
Insert size
| Mean | 4,852.43 |
| Standard Deviation | 98,947.76 |
| P25/Median/P75 | 121 / 143 / 172 |
Mismatches and indels
| General error rate | 0.01% |
| Mismatches | 3,601,348 |
| Insertions | 4,754 |
| Mapped reads with at least one insertion | 0.02% |
| Deletions | 57,171 |
| Mapped reads with at least one deletion | 0.19% |
| Homopolymer indels | 56.8% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| AL123456.3 | 4411532 | 56222600368 | 12,744.4616 | 29,737.1205 |
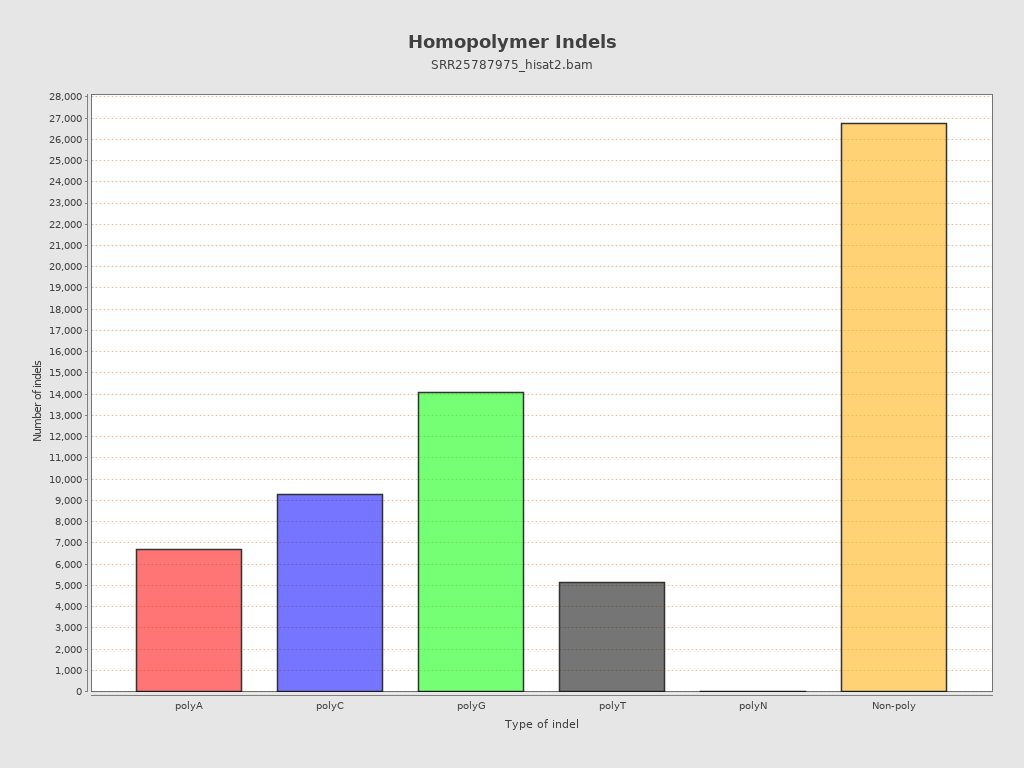
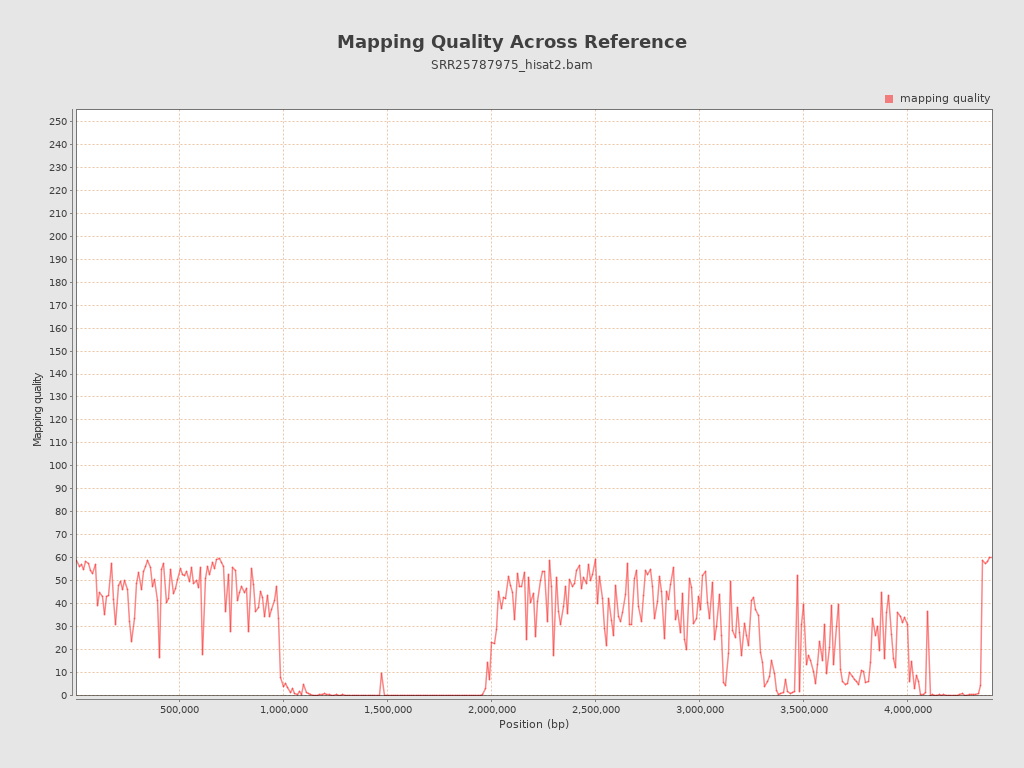
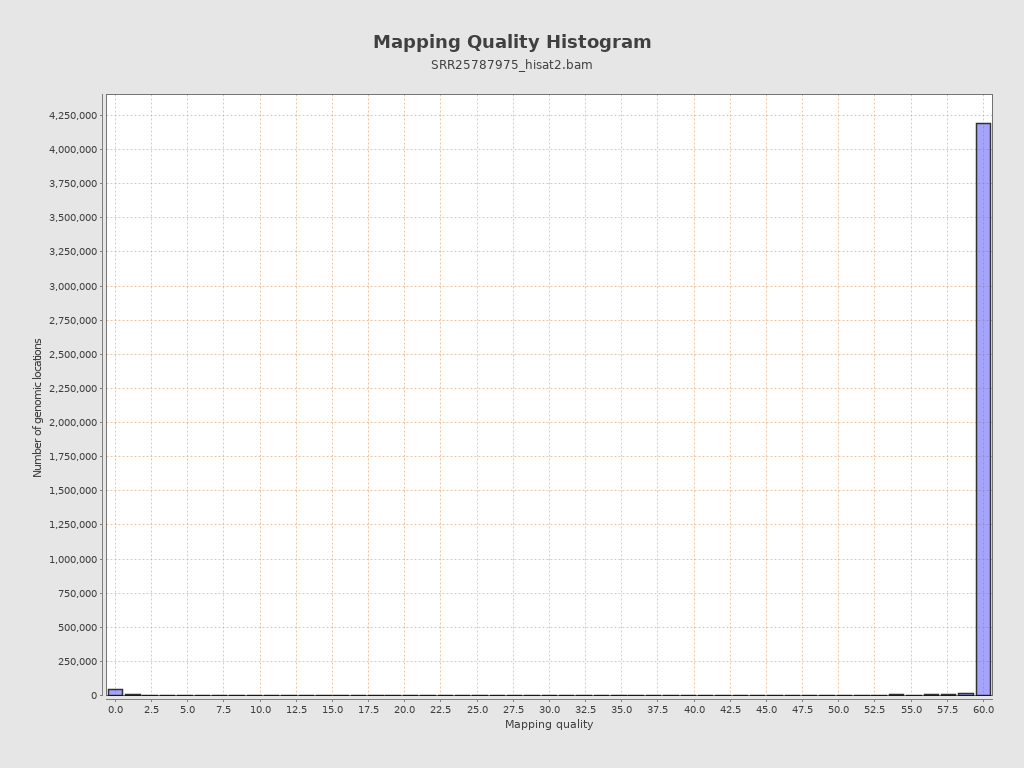
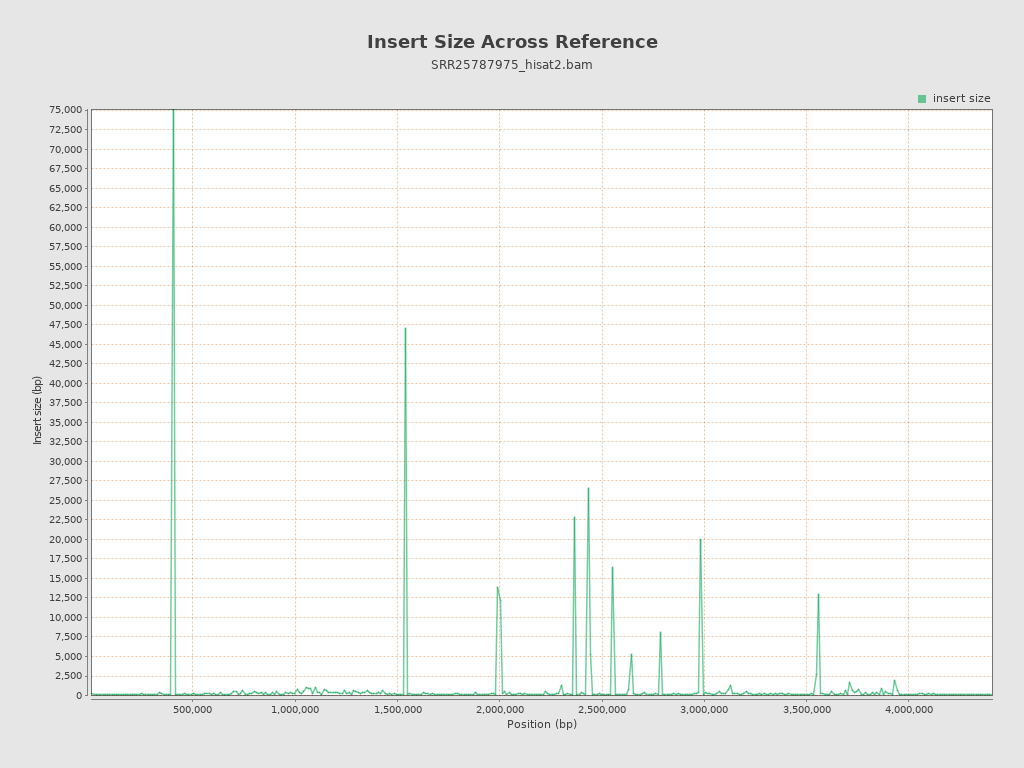
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}